

Molekulare Diagnostik: Methoden
Molekulare Diagnostikmethoden umfassen eine Vielzahl von Techniken, darunter die Sanger-Sequenzierung, Multiplex Ligation-dependent Probe Amplification (MLPA), Array Comparative Genomic Hybridization (Array-CGH) und vor allem das Next-Generation Sequencing (NGS). Die Sanger-Sequenzierung war ein Pionier in der DNA-Sequenzierung, wird aber zunehmend durch NGS ersetzt, was eine schnellere und kostengünstigere Hochdurchsatz-Sequenzierung ermöglicht. NGS revolutioniert die Genomik durch die gleichzeitige Sequenzierung von Millionen von DNA-Fragmenten, was eine umfassende Analyse des Genoms, Identifizierung von krankheitsverursachenden Mutationen und personalisierte Medizin ermöglicht. MLPA ist eine wichtige Methode zur Detektion von Genmutationen und Kopienzahlvariationen, während Array CGH verwendet wird, um chromosomale Aberrationen aufzudecken. Diese fortgeschrittenen Technologien bieten eine höhere Auflösung, Sensitivität und Durchsatzrate im Vergleich zu traditionellen Methoden und haben das Potenzial, die Diagnose und Behandlung von Krankheiten zu verbessern.
Sanger Sequenzierung
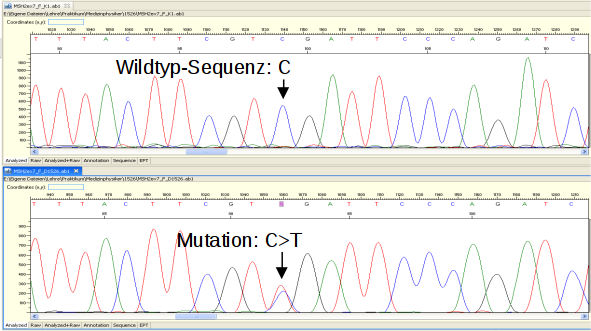
Die Sanger-Sequenzierung ist eine traditionelle Methode zur DNA-Sequenzierung, bei der in der Kettenabbruchreaktion einzelne DNA-Fragmente durch DNA-Polymerase synthetisiert und mit fluoreszierenden Markern markiert werden. Durch die Trennung der Fragmente nach ihrer Größe können die Nukleotidsequenzen bestimmt werden.
In der Diagnostik wird die Sanger-Sequenzierung häufig zur spezifischen Identifizierung von genetischen Mutationen und zur Diagnose von Krankheiten eingesetzt. Sie ermöglicht die Detektion von einzelnen Nukleotidvariationen, die mit erblichen Erkrankungen und Krebs in Verbindung stehen.
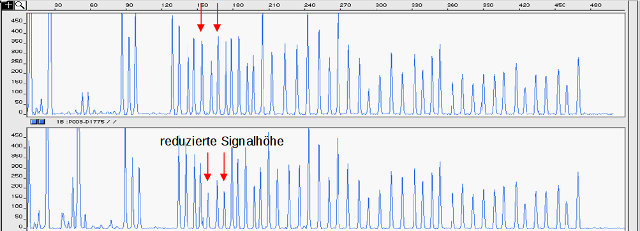
MLPA-Analyse („Multiplex Ligation-dependent Probe Amplification“)
MLPA (Multiplex Ligation-dependent Probe Amplification) ist eine molekulargenetische Methode zur Detektion von Kopienzahlvariationen (CNVs) und zur Quantifizierung von Genabschnitten. Dabei werden spezifische Sonden verwendet, die sich an bestimmte DNA-Sequenzen binden. Durch Verknüpfung und Amplifikation der Sonden können Genkopien quantifiziert und Veränderungen in der Genomkopienzahl identifiziert werden.
In der Diagnostik wird MLPA häufig eingesetzt, um genetische Krankheiten zu diagnostizieren, die durch CNVs verursacht werden, wie zum Beispiel das Prader-Willi-Syndrom oder das Angelman-Syndrom; zusätzlich kann MLPA auch zur Untersuchung von Methylierungszuständen eingesetzt werden. Es ermöglicht auch die präzise Bestimmung von Genkopien in Gewebe von Tumoren, was für die Prognose und Behandlung von Krebspatienten wichtig ist.
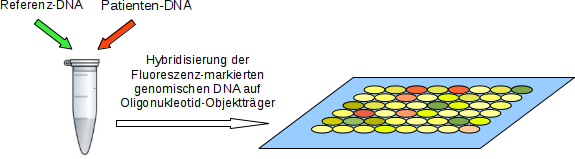
Array-CGH
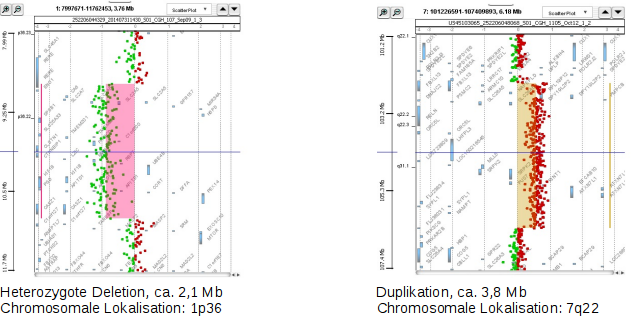
Array Comparative Genomic Hybridization (Array-CGH) ist eine molekulargenetische Technik, die verwendet wird, um genomische Aberrationen, wie zum Beispiel Deletionen, Duplikationen und komplexere chromosomale Rearrangements, zu identifizieren. Dabei werden genomische DNA-Proben des Patienten und Referenz-DNA-Proben auf einem Mikroarray hybridisiert und verglichen. Abweichungen im Signalintensitätsverhältnis zwischen den Patienten- und Referenzproben zeigen genomische Aberrationen an.
In der Diagnostik wird die Array-CGH eingesetzt, um genetische Ursachen von Entwicklungsstörungen, geistigen Behinderungen und angeborenen Anomalien zu identifizieren. Es ermöglicht auch die Charakterisierung von Tumoren und die Bestimmung von prognostischen Markerprofilen für Krebspatienten.
NGS-Sequenzierung („Next-generation sequencing“)
Next-Generation-Sequencing (NGS) bezeichnet eine Gruppe von Hochdurchsatzsequenzierungstechnologien, die eine schnelle und kostengünstige Analyse von DNA ermöglichen. Diese moderne Technologie ermöglicht eine parallele Sequenzierung von Millionen von DNA-Fragmenten. Nach der Sequenzierung erfolgt eine umfangreiche Datenanalyse der Sequenzdaten, mit leistungsfähigen Computeralgorithmen analysiert werden. Die Sequenzen werden mit Referenzgenomen verglichen, um krankheitsassoziierte Varianten (Mutationen) zu identifizieren. Bioinformatische Werkzeuge werden verwendet, um die biologischen und klinischen Implikationen dieser Varianten zu interpretieren. Danach werden die identifizierten genetischen Varianten hinsichtlich ihrer klinischen Relevanz bewertet und ermöglicht, die Verbindung zu genetischen Erkrankungen herzustellen.
In der Panel-Sequenzierung werden zielgerichtet bestimmte Gene oder Regionen des Genoms, die mit bestimmten Krankheiten in Verbindung stehen, sequenziert und analysiert.
Genomweite Sequenzierungen sind nicht zielgerichtet auf bestimmte Krankheiten.
Bei der Exom-Sequenzierung (‚whole exome sequencing‘) wird das Exom, alle Abschnitte der DNA, die für Proteine kodieren, sequenziert. Das Exom umfasst ca. 1-2% des gesamten Genoms bzw. ca. 23.000 Gene. Die Sequenzierung des gesamten Genoms (‚whole genome sequencing‘) erfolgt ungerichtet, so dass auch nicht-kodierende Bereiche erfasst werden.
Genomweite Sequenzierungen mittels NGS, insbesondere Exom- und neuerdings auch die Genom-Sequenzierung, haben die genetische Diagnostik in den letzten Jahren revolutioniert. Durch die Analyse des gesamten Exoms oder Genoms können Wissenschaftler und Ärzte genetische Ursachen von Krankheiten wie Krebs, genetischen Syndromen und seltenen genetischen Störungen identifizieren. Dies ermöglicht eine präzisere Diagnose und personalisierte Behandlungsansätze.
In der klinischen Praxis wird die Exom- und die Genom-Sequenzierung zunehmend eingesetzt, um Patienten mit unklaren klinischen Auffälligkeiten zu diagnostizieren, bei denen herkömmliche diagnostische Methoden keine eindeutige Ursache liefern.
Insbesondere kann eine genomweite Sequenzierung helfen, die genetische Ursache einer seltenen Erkrankung zu identifizieren. Die Identifizierung spezifischer genetischer Varianten kann dann zu maßgeschneiderten Behandlungsansätzen führen und den Einsatz von Therapien ermöglichen, die auf die individuellen genetischen Profile der Patienten abgestimmt sind. Dies ist besonders relevant in der Onkologie. Des Weiteren können die Erkenntnisse über genetische Risiken betroffenen Familien helfen, informierte Entscheidungen über zukünftige Schwangerschaften zu treffen. Die Ergebnisse der Genomsequenzierung können ebenso genutzt werden, um das Risiko für erbliche Krankheiten zu beurteilen, so dassPatienten personalisierte Entscheidungen in Bezug auf Vorsorgeuntersuchungen und Lebensstiländerungen treffen können.
Die genomweite Sequenzierung hat das Potenzial, nicht nur die Kenntnisse der molekularen Genetik revolutionär zu verbessern, sondern auch erhebliche Auswirkungen auf die klinische Praxis, Forschung und öffentliche Gesundheit zu haben. Der Einsatz von künstlicher Intelligenz und maschinellem Lernen verbessert bereits die Variantendetektion, Klassifikation und Priorisierung sowie die Bewertung genetischer Daten.
Auch an unserem Institut nutzen wir Erkenntnisse aus der Genomsequenzierung, um neue Gene und deren Funktionen und entsprechende Signalwege zu identifizieren, um das Verständnis grundlegender biologischer Prozesse vertiefen zu können.