

mtDNA, Quality Control and Aging
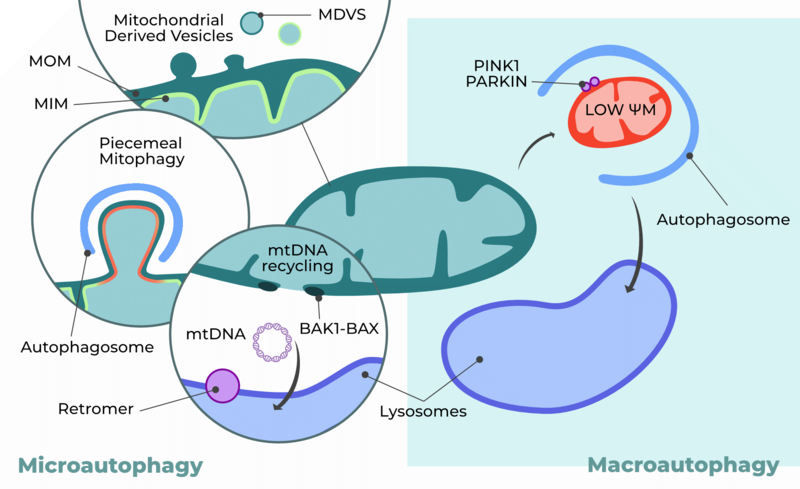
Mitochondrial quality control. In macroautophagy, mitochondrial depolarization triggers the recruitment of specific proteins, such as PINK1 and PARKIN, to the mitochondrial membrane. This initiates the formation and engulfment of the entire mitochondrion into an autophagosome, which subsequently fuses with the lysosome for degradation and recycling. In contrast, microautophagy selectively targets specific mitochondrial compartments or proteins for removal. This category includes pathways such as Mitochondria-Derived Vesicles (MDVs), piecemeal mitophagy, and the recently described selective removal of mitochondrial DNA (mtDNA).
Mitochondria are multifaceted organelles involved in many critical cellular functions essential for cellular homeostasis. In contrast to their traditional view as static energy-producing organelles, mitochondria form a highly dynamic network that is constantly remodeled, adapting its function in real time to cellular needs. Uniquely among organelles, mitochondria contain their own genome, mitochondrial DNA (mtDNA), which encodes 13 protein-coding genes of the oxidative phosphorylation (OXPHOS) machinery. Preserving both the structural and genetic integrity of mitochondria is therefore essential for sustained mitochondrial and cellular function.
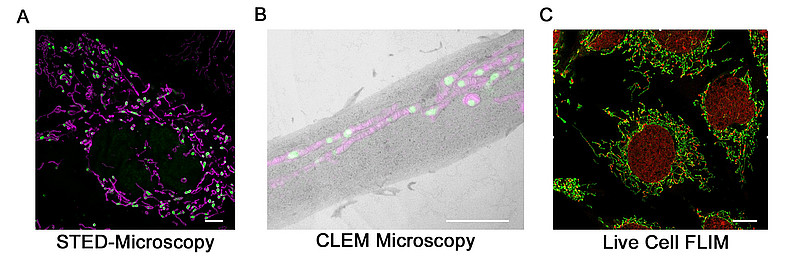
Advanced imaging approaches used in our group to investigate mitochondrial biology. A) STED super-resolution microscopy for nanoscale visualization of mitochondrial organization. B) Correlative Light and Electron Microscopy (CLEM), combining fluorescence and ultrastructural information to precisely localize mitochondrial features. C) Live-cell FLIM microscopy to monitor mitochondrial physiology and metabolic state in living cells. Together, these approaches provide high-resolution structural and functional insights into mitochondrial organization in health and disease.
Cells have evolved a variety of quality control mechanisms that preserve the integrity of proteins, membranes, and even entire organelles. Mitophagy, the selective removal of mitochondria through autophagy, encompasses a group of processes responsible for eliminating dysfunctional organelles, but also specifically damaged mitochondrial components, including the mtDNA. Alterations in the molecular players of these quality control pathways have been linked to many different types of diseases, including metabolic syndromes and neurological disorders, as well as to the natural process of aging.
Our group aims to understand from a molecular perspective, why disease occurs when these systems fail. Our work can be described in four main research lines:
1. Selective degradation of the mtDNA: We investigate the molecular mechanisms responsible for the recognition, trafficking, and selective elimination of damaged mitochondrial DNA, with a particular focus on the interplay between mitochondria, endosomes, and lysosomes in maintaining mtDNA integrity.
2. Pathophysiological mechanisms of Mitochondrial related Charcot-Marie Tooth: Using cellular models, we study how alterations in mitochondrial and lysosomal proteins contribute to peripheral nerve degeneration.
3. mtDNA instability and cellular integrity: We aim to understand how defects in mtDNA replication and maintenance influence cellular homeostasis, stress responses, and tissue function, with special focus on metabolism, aging and neurodegeneration.
4. Mitochondrial adaptations in Lymphoma cells: We investigate how lymphoma cells remodel mitochondrial metabolism to support proliferation, survival, and adaptation to metabolic stress, with the goal of identifying novel therapeutic vulnerabilities.
Interested in joining our lab?
We are always looking for highly motivated students, PhD candidates, postdoctoral researchers, and visiting scientists who share our interest in mitochondrial biology. If you are interested in joining our team, please do not hesitate to contact us.