

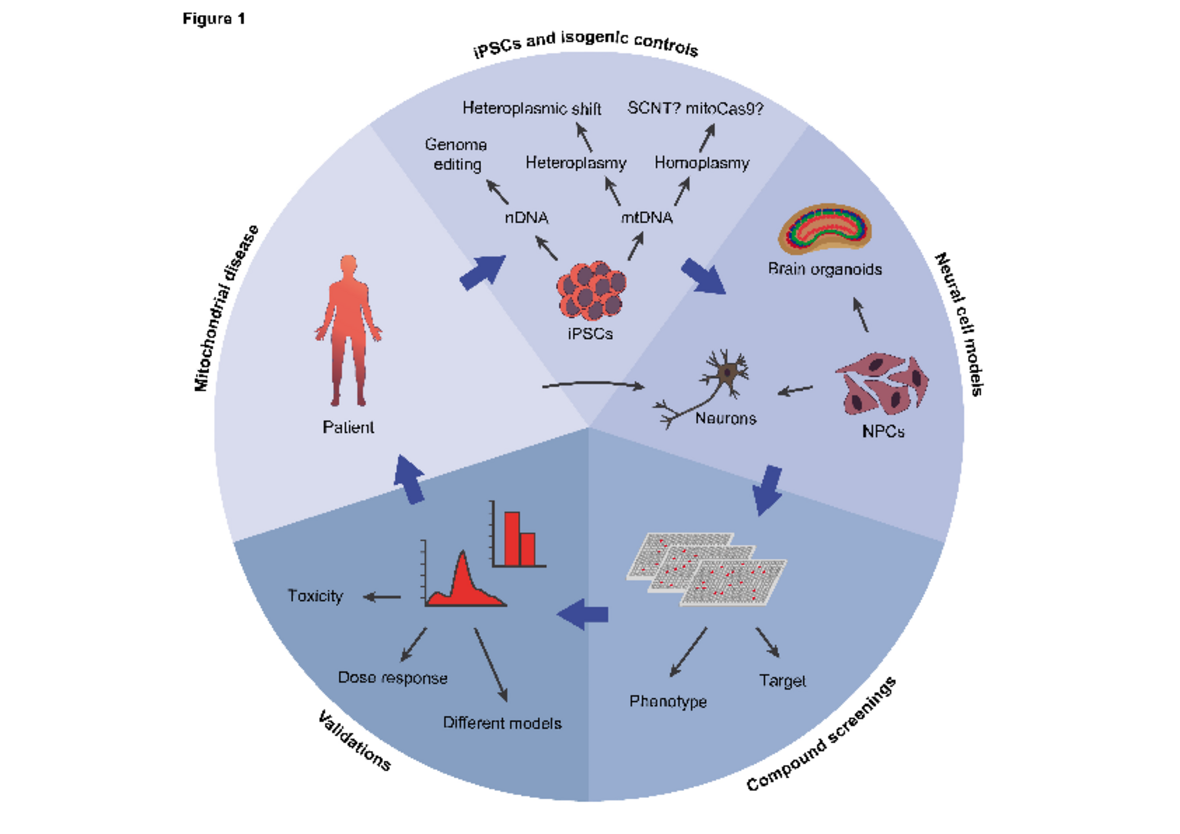
Leigh Syndrom
Das Leigh-Syndrom ist eine seltene Erkrankung bei Kindern und gleichzeitig die schwerste Form im Bereich der mitochondrialen Erkrankungen. Es wird durch Mutationen in Genen verursacht, die sich im nuklearen Genom (NDUFS4 oder SURF1) oder im mitochondrialen Genom (MT-ND6 oder MT-ATP6) befinden. Die Krankheit betrifft speziell das zentrale Nervensystem und verursacht Störungen sowohl im geistigen als auch im motorischen Bereich. Derzeit sind noch keine Heilmittel oder Behandlungen verfügbar.
Unsere Forschungsergebnisse zeigen, dass neurale Vorläuferzellen aus iPSCs, die Mutationen im MT-ATP6 tragen, Fehlfunktionen in der mitochondrialen Calciumhomöostase aufweisen. Diese Fehlfunktion konnte in vitro unter Verwendung von Phosphodiesterase 5-Inhibitoren kompensiert werden (Lorenz et al, Cell Stem Cell 2017). Wir haben festgestellt, dass SURF1-Mutationen die Neurogenese beeinträchtigen, indem sie die frühe Morphogenese stören (Inak et al, BioRxiv 2019).
Leigh Syndrome
Leigh syndrome is a rare disorder and the most severe form of mitochondrial disease in children. It is caused by genetic mutations in genes encoded in the nuclear genome (such as NDUFS4 or SURF1) or in the mitochondrial genome (such as MT-ND6 or MT-ATP6). The disease specifically affects the central nervous system, causing intellectual and movement disturbances. There are currently no cure or treatments available.
We found that iPSC-derived neural progenitors carrying MT-ATP6 mutations exhibit defects in mitochondrial calcium homeostasis that can be reverted in vitro using phosphodiesterase 5 inhibitors (Lorenz et al, Cell Stem Cell 2017). We determined that SURF1 mutations impair the neurogenesis process by disrupting early morphogenesis (Inak et al, BioRxiv 2019).
Juvenile Huntington-Krankheit
Die Huntington-Krankheit (HD) ist eine seltene, unheilbare, neurodegenerative Erkrankung, die durch abnormale CAG-Wiederholungen im Gen Huntingtin (HTT) verursacht wird. Bei Personen mit über 60 Wiederholungen treten die Krankheitssymptome bereits im Kindesalter auf und verursachen eine Krankheit, die als Juvenile Huntington-Krankheit (JHD) bekannt ist. Ein möglicher Grund für den fortschreitenden Verlust von Neuronen bei Huntington und JHD könnte die abnormale Mitochondrienfunktion sein (Cunnane et al., Nature Review Drug Discovery 2020). Mitochondriale Defekte können früh im Krankheitsverlauf auftreten, welche möglicherweise zu einem Kompensationsverlust in den Neuronen führen und folglich absterben.
Wir verwenden patientenspezifische iPSCs und Genom-Engineering, um die Rolle der mitochondrialen Beeinträchtigung bei JHD zu untersuchen und mögliche Behandlungsstrategien zu identifizieren (Scior et al., EMBO J, 2018).
Juvenile Huntington's disease
Huntington’s disease (HD) is a rare incurable neurodegenerative disorder caused by an abnormal CAG repeats in the gene Huntingtin (HTT). In individuals with repeats higher than 60, the disease symptoms appear during childhood and cause a disease defined Juvenile Huntington´s disease (JHD). The specific progressive loss of neurons in HD and JHD may be a consequence of the neuronal dependence on mitochondrial function (Cunnane et al, Nature Review Drug Discovery 2020). Mitochondrial defects may occur early in the disease process and may contribute to the decompensation of neurons and their ultimate loss.
We are using patient specific iPSCs and genome engineering to dissect the role of mitochondrial impairment in JHD to possibly identify counteracting strategies (Scior et al, EMBO J, 2018).